Getting Started¶
Here you will learn how to run your first FragPELE’s simulation.
How to do it?¶
Previous Requisites¶
- Complex PDB: PDB processed, with the protein and the ligand prepared to run with PELE. Remember to rename the ligand chain to “L”.
- Fragment PDB: PDB with the desired fragment. Remember to rename the chain to “L”.
- Serie file: The instructions that explain how the growing has to be done are stored in this file. It must have the format described bellow.
Growing of a fragment to a core¶
Grow 1 fragment to 1 cores
| fragment PDB 1 | heavy atom core 1 | heavy atom fragment 1 |
ie:
amino.pdb C1 N1
Growing of two fragment to the same core¶
Add 1 fragment to an initial core. Then, add another fragment to the same core on the output of the 1st growing. Example: Grow two radicals onto a phenyl
| fragment PDB 1 | heavy atom core 1 (C1) | heavy atom fragment 1 | fragment PDB 2 | heavy atom core 1 (C2) | heavy atom fragment 2 |
i.e:
amino.pdb C1 N1 phenyl C1 C1
Growing of a fragment to a previously grown fragment¶
Add 1 fragment to an initial core. Then, add another fragment to the previously grown fragment. Example: Grow one radical onto a phenyl and then another fragment into the previously grown radical.
| fragment 1 | heavy atom core 1 | heavy atom fragment 1 | fragment PDB 2 | fragment atom core 1 (C2)*fragment_number* | heavy atom fragment 2 |
i.e:
amino.pdb C1 N1 phenyl.pdb N1*1* C1
All together¶
Add 2 fragments onto different position of the same core. Then, perform another growing with a 3rd fragment onto a different core and a 4th into the previosuly grown fragment.
| fragment PDB 1 | heavy atom core 1 (C1) | heavy atom fragment 1 | fragment PDB 2 | heavy atom core 1 (C2) | heavy atom fragment 2 |
| fragment PDB 3 | heavy atom core 2 | heavy atom fragment 3 | fragment PDB 4 | heavy atom fragment 3 | heavy atom fragmnet 4 |
i.e:
amino.pdb C1 N1 phenyl C3 C1
ethyl C1 C2 methyl C1*1* C1
Launch simple FrAG-PELE simulation¶
Default: 10 growing steps of 6 pele steps, and a final equilibration simulation of 20 pele steps
python3.X main.py -cp path_to_complex_pdb -sef serie_file
Growing a fragment specifying direction¶
FragPELE selects by default an H atom with less clashes with the protein. It will take this atom as direction to grow, replacing this hydrogen by the whole fragment. Additionally, the atom of the fragment that will be replaced is also selected randomly. Sometimes you would like to specify which atom to replace. Then, the format to do that is the following:
| fragment PDB 1 | heavy atom core 1 - H atom core | heavy atom fragment 1 - H atom fragment |
i.e:
amino.pdb C1-H1 N1-H1
In FragPELE 2.0 can also grow fragments onto NON-hydrogen atoms, but you must specify the atom name:
amino.pdb C1-BR1 N1-H1 (BR1 is a heavy atom, therefore, it can not be automatically detected and must be specified)
Growing through BOND-LIKE selection¶
In FragPELE 2.0, you can also grow through a BOND-LIKE selection.
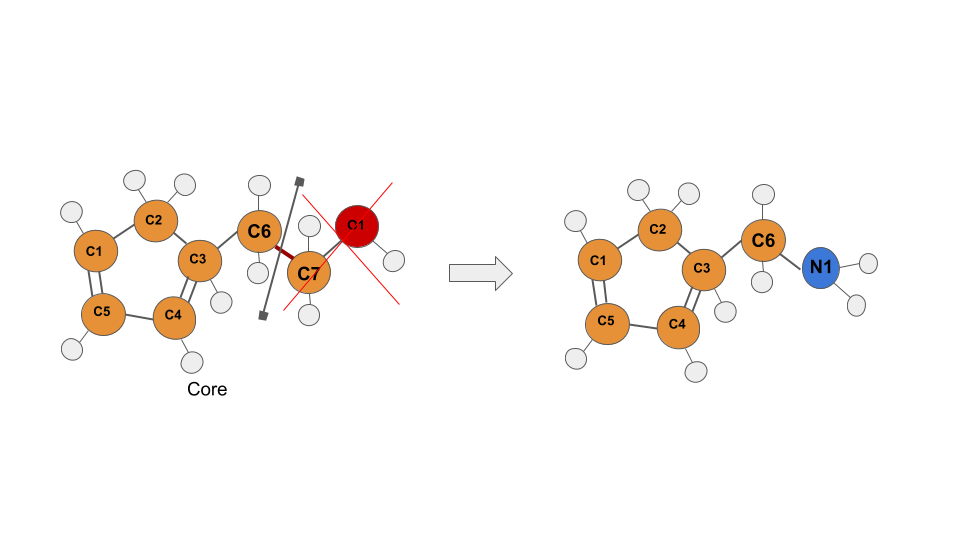
In this case, as the selection has been done in a NON-terminal bond, the tree of atoms after the selected bond will be deleted and replaced by the fragment.
i.e:
amino.pdb C6-C7 N1-H1
Growing by double or triple bonding¶
Imagine that we have the following situation:
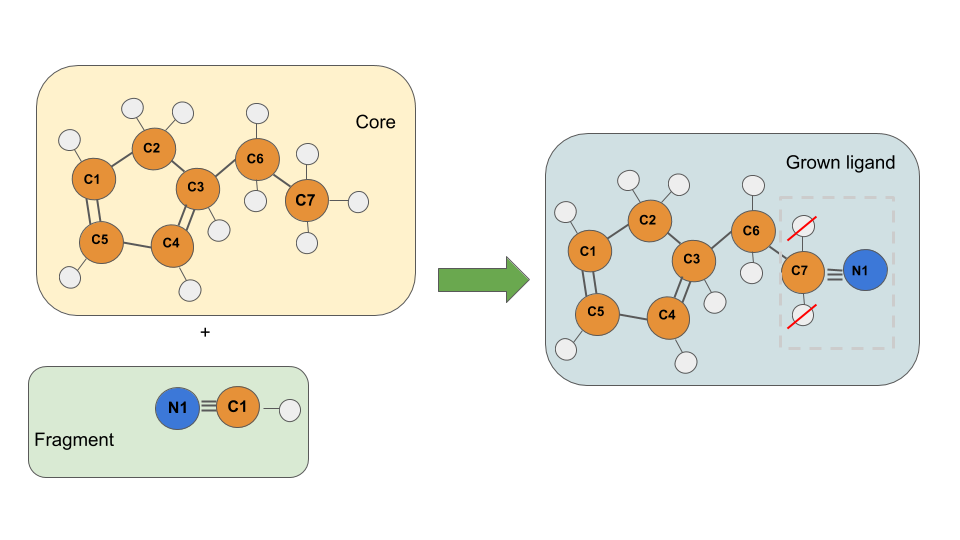
In this case we would like to replace a simple bond by a triple bond, thus, it is required to have a fragment containing this triple bond (at least the fragment must have two heavy atoms, otherwise would never have valid double or triple bonds).
The software automatically will detect which bond type is present between the selected atoms of the fragment. So, in the input you just need to specify the atom names of this atoms:
ciano.pdb C7-H11 N1-C1
Take in mind that the “BOND direction” defined by (first_atom - second_atom) also defines the direction of atom’s deletion, so, in the fragment’s case writing the N1-C1 bond are keeping the N1 and remove everything towards.